UMJETNA INTELIGENCIJA STOJI IZA SVEGA Softver umjetne inteligencije gotovo je predvidio omikronovu strukturu
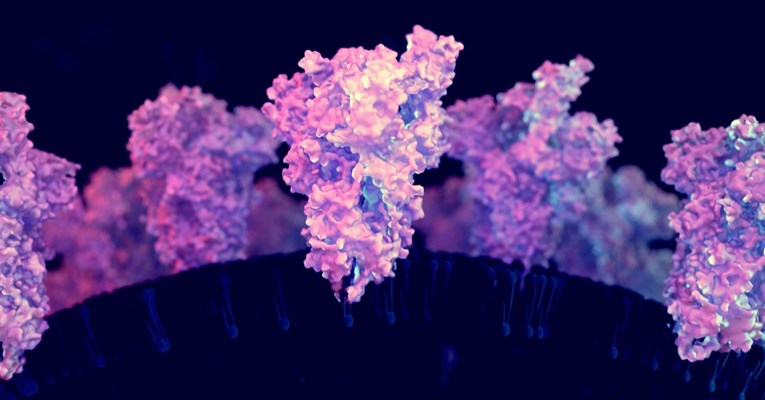
SVJETSKA zdravstvena organizacija (WHO) označila je 26. studenoga soj koronavirusa koji se pojavio u Južnoj Africi “zabrinjavajućom varijantom” i nazvala ga je omikron. Sljedećeg dana profesor sa Sveučilišta British Columbia u Kanadi Sriram Subramaniam preuzeo je sekvencu genoma objavljenu na internetu i dogovorio da se uzorci omikronova DNK pošalju u njegov laboratorij.
Subramaniamova znanstvena ekipa koristi elektronske mikroskope kako bi otkrila 3D strukturu proteina i bolje razumjela njihov rad. Već je mapirao proteine šiljka koji koronavirusi koriste za vezanje i ulazak u ljudske stanice za neke ranije sojeve. Opisivanje omikronovog proteina šiljka činilo se hitnim jer se njegov DNK razlikovao, što je moglo objasniti brzo širenje ove varijante.
No, kao i drugi koji su tog vikenda kupovali putem interneta, Subramaniam je morao biti strpljiv: dok DNK nije stigao poštom, nije mogao staviti omikronove proteine pod mikroskop, piše Wired.
Umjetnom inteligencijom nakon sat vremena dobio rezultate
S druge strane američkog kontinenta, Colby Ford, istraživač računalne genomike na Sveučilištu Sjeverne Karoline u Charlotteu, također je razmišljao o omikronovu proteinu šiljku. Rodbina mu je postavljala pitanje koje je također zabrinjavalo mnoge stručnjake – bi li omikron mogao izbjeći postojeća cjepiva? Ta cjepiva uče tijelo da reagira na proteine šiljka prijašnjih sojeva.

Umjesto naručivanja laboratorijskih potrepština, Ford je isprobao nedavno izmišljeni prečac. Istog dana kada je WHO dao omikronu ime, koristio je besplatni softver umjetne inteligencije kako bi pokušao predvidjeti strukturu iz slijeda aminokiselina kodiranih u omikronovom genomu.

Za otprilike sat vremena Ford je dobio prve rezultate i brzo ih objavio na internetu. Početkom prosinca on i dvojica kolega objavili su potpuniji rad, koji je sada prihvaćen za objavljivanje, uključujući predviđanja da će neka antitijela na prethodne sojeve biti manje učinkovita protiv omikrona.
Subramaniamov laboratorij ubrzo je objavio svoja mikroskopska promatranja strukture zajedno s rezultatima testova antitijela 21. prosinca. Jedna od dvije Fordove predviđene strukture pokazala se prilično točnom, izračunao je da se položaji njegovih središnjih atoma razlikuju za oko pola angstrema, oko polumjera vodikovog atoma.
Neki istraživači su sumnjali u softver
Način na koji su predviđanja išla ispred eksperimenata odražava nedavnu promjenu u molekularnoj biologiji koju je donijela umjetna inteligencija. Prvi softver sposoban točno predvidjeti proteinske strukture postao je široko dostupan samo nekoliko mjeseci prije nego što se omikron pojavio.
Fordovi su rezultati bili sugestivniji nego konačni, jer softver nije bio dizajniran ili potvrđen za predviđanje malih promjena uzrokovanih mutacijama poput onih u omikronu. Neki istraživači su se prema njima odnosili sa sumnjom.
Budući da oblik proteina određuje njegovo ponašanje, poznavanje njegove strukture može pomoći svim vrstama bioloških istraživanja, od studija evolucije do rada na bolestima. U istraživanju lijekova otkrivanje strukture proteina može pomoći u otkrivanju potencijalnih ciljeva za nove tretmane.
Dešifriranje strukture proteina tradicionalno je uključivalo mukotrpan laboratorijski rad. Većina od otprilike 200.000 poznatih struktura mapirana je pomoću procesa u kojem se proteini formiraju u kristal i bombardiraju rendgenskim zrakama. Novije tehnike, poput elektronske mikroskopije koju koristi Subramaniam, mogu biti brže, ali proces je još uvijek daleko od jednostavnog.

Svijet je imao dva načina za predviđanje strukture proteina
Krajem 2020. godine dugogodišnja nada da bi računala mogla predvidjeti strukturu proteina iz sekvence aminokiselina odjednom je postala stvarnost, nakon desetljeća sporog napretka. DeepMind softver pod nazivom AlphaFold pokazao se toliko točnim u predviđanju proteina da je suosnivač John Moult, profesor na Sveučilištu u Marylandu, proglasio problem riješenim.
David Baker, čiji laboratorij na Sveučilištu Washington radi na predviđanju strukture proteina, koristio je tragove koje je izbacio DeepMind kako bi usmjerio dizajn softvera otvorenog koda pod nazivom RoseTTAFold. Bio sličan, ali ne tako moćan kao AlphaFold. Oba se temelje na algoritmima strojnog učenja koji su izoštreni za predviđanje proteinskih struktura treniranjem na kolekciji od više od 100.000 poznatih struktura. Odjednom, svijet je imao dva načina za predviđanje strukture proteina.
Minkyung Baek, postdoktorska istraživačica u Bakerovom laboratoriju koja je vodila rad na RoseTTAFoldu, kaže da se predviđanja nisu pokazala ključnima za rad na covidu-19, ali i da vjeruje da će postati sve važnija za odgovor na buduće bolesti. Ona sada pokušava navesti RoseTTAFold da točno predvidi strukturu antitijela i invazivnih proteina kada su povezani, što bi softver učinilo korisnijim za projekte zaraznih bolesti.
Unatoč impresivnoj izvedbi, prediktori proteina ne otkrivaju sve o molekuli. Izbacuju jednu statičnu strukturu za protein i ne hvataju promjene koje se događaju kada je u interakciji s drugim molekulama. Algoritmi su obučeni na bazama podataka poznatih struktura, koje više odražavaju one koje je najlakše eksperimentalno mapirati, a ne punu raznolikost prirode.
Ova kombinacija objašnjava zašto omikron može preplaviti čak i visoko cijepljene zajednice
Osim strukture proteina šiljka, Subramaniamov rad također je uključivao rezultate koje umjetna inteligencija još nije svladala – kombiniranu strukturu šiljka vezanog za ljudski protein na koji cilja. Rezultati sugeriraju da strukturne promjene omikrona omogućuju da se jače veže na stanice domaćina, a istovremeno je manje ranjiv na antitijela iz prethodnih sojeva.
Ova kombinacija objašnjava zašto omikron može preplaviti čak i visoko cijepljene zajednice, zaključuje Wired.

INDEX.HR